×

我们的网站(https://second.b2star.com)与 谷歌 Chrome 和 Safari 等浏览器完全兼容。 为了获得最佳浏览体验,我们建议使用以上这些浏览器。
您可能遇到了一些问题:
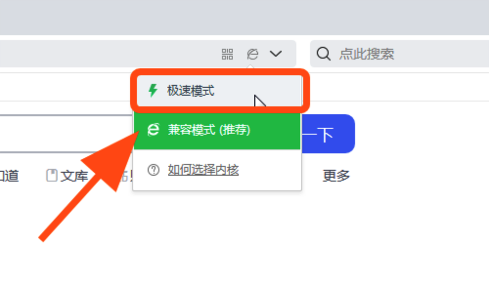
如果您使用的是 QQ 浏览器 或 360 浏览器,请注意某些页面可能无法正确显示,或者某些文本可能会丢失。要解决此问题,请确保在浏览器设置中切换到极速模式。
如要解决该问题,请将浏览模式切换为 “极速模式” ,或使用谷歌Chrome和Safari浏览器进行访问
您可能遇到了一些问题:
如果您使用的是 QQ 浏览器 或 360 浏览器,请注意某些页面可能无法正确显示,或者某些文本可能会丢失。要解决此问题,请确保在浏览器设置中切换到极速模式。
如要解决该问题,请将浏览模式切换为 “极速模式” ,或使用谷歌Chrome和Safari浏览器进行访问